Automatische Übersetzung anzeigen
Dies ist eine automatisch generierte Übersetzung. Wenn Sie auf den englischen Originaltext zugreifen möchten, klicken Sie hier
#Produkttrends
{{{sourceTextContent.title}}}
Wann trat 21 CFR Part 11 in Kraft: Was es ist und wie man die Einhaltung der Vorschriften gewährleistet
{{{sourceTextContent.subTitle}}}
Die Einhaltung der Vorschriften gemäß 21 CFR Part 11 ist für die von der FDA regulierte Pharma- und Nahrungsergänzungsmittelbranche verbindlich vorgeschrieben. Wenn Sie nicht wissen, worum es sich dabei handelt und wie Sie die Einhaltung dieser Vorschriften gewährleisten können, finden Sie in diesem Abschnitt die Antworten auf Ihre Fragen.
{{{sourceTextContent.description}}}
In regulierten Fertigungsbranchen sind digitale Aufzeichnungen längst mehr als nur einfache Dateien zur Speicherung von Daten. Sie sind zu rückverfolgbaren Beweismitteln geworden.
Für Unternehmen aus den Bereichen Pharmazie, Nutrazeutika, Medizinprodukte und Auftragsfertigung sind elektronische Aufzeichnungen und elektronische Signaturen unerlässlich, um die Produktionssicherheit zu gewährleisten, die Qualitätssicherung aufrechtzuerhalten und kostspielige Störungen zu vermeiden, die durch Änderungen der Rezeptur oder Anpassungen der Prozessparameter verursacht werden. Im Zuge der Weiterentwicklung der US-amerikanischen Vorschriften ist die Einhaltung von 21 CFR Part 11 nach und nach von einer optionalen Investition zu einer zwingenden Anforderung geworden.
Für viele Hersteller, insbesondere kleine und mittlere Unternehmen, kann die Implementierung von Systemvalidierungen und konformen Datenmanagementlösungen kostspielig und komplex erscheinen. Daher werden oft zunächst zwei zentrale Fragen gestellt: Wann trat 21 CFR Part 11 in Kraft, und was bedeutet die Einhaltung von 21 CFR Part 11?
Dieser umfassende Leitfaden bietet eine detaillierte Erläuterung von 21 CFR Part 11. Wir werden untersuchen, was 21 CFR Part 11 ist, wer die Vorschriften einhalten muss, welche wesentlichen Anforderungen bestehen und wie die Einhaltung von 21 CFR Part 11 auf praktische und kosteneffiziente Weise erreicht werden kann.
Die wichtigsten Erkenntnisse
Wann trat 21 CFR Part 11 in Kraft und wie sieht der historische Kontext aus?
Was bedeutet 21 CFR Part 11 für die pharmazeutische Industrie?
Wer muss in verschiedenen Branchen die Anforderungen von 21 CFR Part 11 erfüllen?
Kernanforderungen von 21 CFR Part 11 in Bezug auf Aufzeichnungen, Signaturen, Zugriff und Prüfpfade.
Wie man die Anforderungen von 21 CFR Part 11 Schritt für Schritt erfüllt.
Wie Part-11-konforme Geräte die Produktion von Arzneimitteln und Nutrazeutika unterstützen.
1. Wann trat 21 CFR Part 11 in Kraft?
Laut der offiziellen Website der FDA trat 21 CFR Part 11 am 20. August 1997 in Kraft.
Was war der Anlass für den Erlass dieser Verordnung? Ihr Ursprung liegt in dem umfassenden Übergang von papierbasierten Aufzeichnungen zu computergestützten Systemen. Zuvor wurden Produktionsaufzeichnungen, Laborberichte und Qualitätsdokumente in der pharmazeutischen Industrie größtenteils auf Papier erstellt, unterzeichnet und aufbewahrt. Mit zunehmendem Produktionsumfang konnten einfache Papieraufzeichnungen den Anforderungen an eine schnelle Dokumentation und den sofortigen Datenabruf nicht mehr gerecht werden, was Unternehmen dazu veranlasste, digitale Systeme einzuführen.
Um die Zuverlässigkeit dieser neuen Systeme zu gewährleisten, unterwirft Teil 11 des 21 Code of Federal Regulations elektronische Aufzeichnungen und elektronische Signaturen einer strengen behördlichen Aufsicht. Wenn Unternehmen digitale Daten zur Unterstützung von Entscheidungen nutzen, die der FDA-Regulierung unterliegen, müssen diese Daten korrekt und sicher sein sowie bei FDA-Inspektionen rückverfolgbar und abrufbar sein.
2. Was ist 21 CFR Teil 11?
Definition von 21 CFR Teil 11
Was ist 21 CFR Teil 11 im Kern? Es handelt sich um eine von der US-amerikanischen FDA erlassene Vorschrift, die festlegt, wie elektronische Aufzeichnungen und Signaturen zu verwalten sind.
Um die Vorschriften von 21 CFR Part 11 zu verstehen, sollte man sich die frühere Aufsicht der FDA über verschlossene Aktenschränke vor Augen führen, bei der überwacht wurde, wer diese verwaltete, und nachverfolgt wurde, wer ihren Inhalt öffnete oder änderte. Da sich diese Aktenschränke zu digitalen Systemen weiterentwickelt haben, hat die FDA die Richtlinien von 21 CFR Part 11 eingeführt, um sicherzustellen, dass die Systeme ordnungsgemäß kontrolliert werden und die Daten sicher bleiben.
Welche Arten von Aufzeichnungen sind davon betroffen?
Zu den elektronischen Aufzeichnungen gemäß 21 CFR Part 11 können gehören:
Elektronische Chargenprotokolle
Herstellungsaufzeichnungen
Qualitätskontrollaufzeichnungen
Betriebsprotokolle von Geräten
Laboruntersuchungsergebnisse
Reinigungs- und Wartungsaufzeichnungen
Schulungsaufzeichnungen
Aufzeichnungen zur Änderungskontrolle und zu Abweichungen
Es ist zu beachten, dass diese Vorschrift nicht für alle digitalen Dateien gilt; sie gilt nur dann, wenn elektronische Aufzeichnungen verwendet werden, um die Anforderungen der FDA an die Aufbewahrung von Aufzeichnungen zu erfüllen.
Welche Branchen müssen die Vorschriften einhalten?
Die Compliance-Anforderungen gemäß 21 CFR Part 11 können für eine Vielzahl von Unternehmen gelten, darunter Hersteller von Arzneimitteln, Nahrungsergänzungsmitteln und Medizinprodukten sowie Auftragsfertiger, Prüflabore und Unternehmen, die von der FDA regulierte Produkte in die Vereinigten Staaten exportieren.
Daher sind Fragen wie „Gilt 21 CFR Part 11 für Medizinprodukte?“ und „Was umfasst 21 CFR Part 11?“ von entscheidender Bedeutung; Das Kernprinzip, das die Anwendbarkeit der Vorschrift regelt, besagt, dass die Compliance-Anforderungen immer dann greifen, wenn elektronische Systeme zur Unterstützung von Aufzeichnungen oder Genehmigungen eingesetzt werden, die den Anforderungen der FDA an die Datenintegrität unterliegen.
3. Warum wurde 21 CFR Part 11 eingeführt?
21 CFR Part 11 wurde eingeführt, weil die digitale Fertigung sowohl neue Effizienzgewinne als auch neue Compliance-Risiken mit sich brachte.
21 CFR Teil 11 entstand als Reaktion auf die Effizienzgewinne und die neuen Compliance-Risiken, die mit der digitalen Fertigung verbunden sind. Als die Branche von papierbasierten Aufzeichnungen auf digitale Systeme umstellte, musste die FDA Vorschriften erlassen, um die Zuverlässigkeit elektronischer Aufzeichnungen zu gewährleisten; diese Vorschrift gilt für von der FDA regulierte Aufzeichnungen, die elektronisch erstellt, geändert, gepflegt, archiviert, abgerufen oder übertragen werden.
Das Kernziel der Vorschrift besteht darin, die Datenintegrität gemäß 21 CFR Part 11 zu gewährleisten und Manipulationen oder Fälschungen zu verhindern, indem Systeme mit Prüfpfaden ausgestattet sein müssen, die ähnlich wie eine „Black Box“ die tatsächliche Abfolge von Ereignissen aufzeichnen. Bei FDA-Inspektionen prüfen die Aufsichtsbehörden elektronische Chargenprotokolle, die Anforderungen an Prüfpfade gemäß Teil 11, Benutzerzugriffskontrollen sowie elektronische Signaturen, die den Anforderungen von 21 CFR Teil 11 entsprechen; diese umfassenden historischen Aufzeichnungen bilden den Grundstein für die Einhaltung gesetzlicher Vorschriften, die Produktqualität und das Vertrauen der Kunden.
4. Wer muss die Anforderungen von 21 CFR Teil 11 erfüllen?
Nicht jedes Unternehmen benötigt das gleiche Maß an Kontrolle, doch viele von der FDA regulierte Hersteller sollten anhand ihrer Systeme und ihres Marktes prüfen, wer die Einhaltung von 21 CFR Part 11 benötigt.
Arzneimittelhersteller
In der pharmazeutischen Industrie gemäß 21 CFR Part 11 müssen elektronische Aufzeichnungssysteme, die Chargeninformationen, Qualitätskontrolle, Anlagenprotokolle oder Freigabegenehmigungen umfassen, in der Regel den Anforderungen von 21 CFR Part 11 entsprechen. Sobald elektronische Aufzeichnungen jedoch Papierunterlagen offiziell ersetzen, wird diese Compliance-Verpflichtung verbindlich und ist nicht mehr optional.
Hersteller von Nahrungsergänzungsmitteln
Wenn ein Hersteller von Nahrungsergänzungsmitteln Produktions-, Qualitäts- oder Chargenprotokolle elektronisch verarbeitet und das Produkt für den Verkauf in den Vereinigten Staaten bestimmt ist, gilt wahrscheinlich die Konformität mit CFR Part 11.
Hersteller von Medizinprodukten
Für diejenigen, die fragen, ob 21 CFR Part 11 für Medizinprodukte gilt: Die Antwort lautet „Ja“, wenn die elektronischen Aufzeichnungen die Anforderungen des FDA-Qualitätssystems erfüllen.
Auftragsfertiger (CMOs)
Auftragsfertiger (CMOs) benötigen oft eine strenge Einhaltung von FDA CFR Part 11, da sie mehrere Kunden bedienen und häufigen Kundenaudits unterliegen.
Unternehmen, die in die Vereinigten Staaten exportieren
Selbst wenn der Hersteller seinen Sitz außerhalb der Vereinigten Staaten hat, ist die Einhaltung der Anforderungen von Teil 11 in der Regel dennoch erforderlich, sofern die Produkte in den USA verkauft werden und die elektronischen Aufzeichnungen sich auf von der FDA regulierte Aktivitäten beziehen.
5. Was sind die Anforderungen von 21 CFR Teil 11?
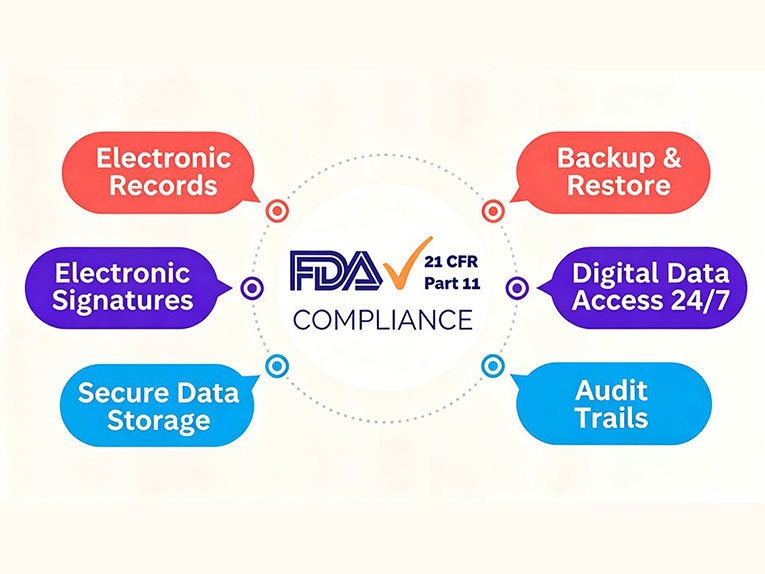
Was genau sind nun laut dem Dokument die Anforderungen von 21 CFR Part 11? Die Antwort lässt sich in mehrere praktische Kontrollmaßnahmen unterteilen. Die Compliance-Anforderungen gemäß 21 CFR Part 11 konzentrieren sich auf fünf Schlüsselbereiche: elektronische Aufzeichnungen, elektronische Signaturen, Prüfpfade, Benutzerzugriffskontrollen sowie Aufbewahrung und Abruf von Aufzeichnungen.
Elektronische Aufzeichnungen
Die Anforderungen von 21 CFR Part 11 an elektronische Aufzeichnungen besagen, dass diese korrekt, vollständig, sicher, lesbar und abrufbar sein müssen. Unternehmen sollten eine ordnungsgemäße Aufbewahrungsfrist für Aufzeichnungen einhalten und für eine sichere Datenspeicherung sorgen.
Elektronische Signaturen
Elektronische Signaturen gemäß FDA Part 11 müssen Identität und Verantwortlichkeit eindeutig nachweisen und den Namen des Unterzeichners, den Zeitstempel sowie die Bedeutung der Aktion (z. B. „Freigabe“) anzeigen, um sicherzustellen, dass sie die gleiche rechtliche Gültigkeit wie handschriftliche Unterschriften besitzen.
Prüfpfade
Eine häufig gestellte Frage ist, wie Prüfpfade bei pharmazeutischen Anlagen funktionieren. Prüfpfade zeichnen automatisch alle kritischen Aktionen auf, darunter Erstellung, Änderung, Löschung, Freigabe und Systemänderungen, um die Rückverfolgbarkeit zu gewährleisten. Wenn die Gewichtsdaten einer Charge von 500 kg auf 510 kg geändert werden, erfasst das Audit-Protokoll den Änderer, den Zeitpunkt der Änderung sowie die Werte vor und nach der Bearbeitung, was die Ursachenanalyse erleichtert.
Zugriffskontrollen für Benutzer
Zugriffskontrollen stellen sicher, dass nur autorisiertes Personal bestimmte Aktionen ausführen kann – durch Passwortschutz und Berechtigungsmanagement unter Verwendung von Multi-Faktor-Authentifizierung und dem Prinzip der geringsten Berechtigungen.
Aufbewahrung und Abruf von Aufzeichnungen
Aufzeichnungen müssen während der gesamten von der FDA vorgeschriebenen Aufbewahrungsfrist jederzeit leicht zugänglich sein; das Versäumnis, Daten während eines Audits umgehend abzurufen, gilt als schwerwiegender Verstoß gegen die Compliance-Vorschriften.
6. Einhaltung von 21 CFR Part 11
Die Einhaltung der Vorschriften ist für die meisten Pharma- und Gesundheitsunternehmen ein vorrangiges Anliegen. In diesem Abschnitt werden wir das Thema eingehend beleuchten und dabei konkrete Umsetzungsschritte, potenzielle Probleme sowie entsprechende Lösungen behandeln.
Schritt 1 – Durchführung einer Konformitätsbewertung gemäß 21 CFR Part 11
Zunächst sollten Sie alle Systeme, die von der FDA regulierte elektronische Aufzeichnungen erstellen, ändern, speichern, abrufen oder übertragen, mit den Richtlinien von 21 CFR Part 11 für Arzneimittel abgleichen. Die Bewertung sollte die Validierung, den Prüfpfad, die Benutzerzugriffskontrolle, elektronische Signaturen, die Aufbewahrung von Aufzeichnungen und die Sicherheit umfassen.
Die Verwendung der Checkliste zur Einhaltung von 21 CFR Teil 11 kann Ihren Prozess beschleunigen.
Schritt 2 – Validierung computergestützter Systeme
Ein 21 CFR Part 11-konformes System sollte einer Validierung unterzogen werden, um nachzuweisen, dass es wie beabsichtigt funktioniert. In der Praxis sollte die Validierung als strukturierte Abfolge von Maßnahmen durchgeführt werden:
1. Systemumfang und Risiken definieren und bewerten
Dokumentieren Sie zunächst klar und deutlich, welche Aufgaben das System erfüllen soll, und identifizieren Sie, wo Risiken bestehen. Beispielsweise muss eine Kapselabfüllmaschine das Füllgewicht, die Geschwindigkeit und die Chargennummer aufzeichnen.
2. Systemleistung testen und verifizieren
Führen Sie anschließend strukturierte Tests mithilfe von IQ, OQ und PQ durch. Diese Bewertung dient dazu, zu überprüfen, ob das System wie erwartet funktioniert.
3. Dokumentieren, genehmigen und kontrollieren
Schließlich müssen alle Validierungsaktivitäten vor der Freigabe des Systems dokumentiert, überprüft und genehmigt werden.
Schritt 3 – Implementierung der Audit-Trail-Funktionalität
Sie müssen die Integrität der Audit-Trail-Funktion sicherstellen und bestätigen, dass das System Daten wie Zeitstempel und Änderungen sicher speichert und aufzeichnet und deren genaue Abfrage ermöglicht. Nehmen wir als Beispiel die Anpassung der Parameter einer Kapselabfüllmaschine: Sobald ein Bediener die Abfüllparameter ändert, erfasst das System automatisch die alten und neuen Werte, die Bediener-ID sowie den Zeitstempel.
Schritt 4 – Kontrollen für elektronische Signaturen einrichten
Um die Anforderungen der FDA an elektronische Signaturen zu erfüllen, sollte jeder Benutzer über eine eindeutige Identität verfügen. Elektronische Signaturen sollten mit bestimmten Datensätzen verknüpft sein und dürfen nicht kopiert oder neu zugewiesen werden.
Schritt 5 – Benutzerzugriff kontrollieren
Verwenden Sie rollenbasierten Zugriff. Verschiedene Rollen sollten unterschiedliche Berechtigungen haben. Administratoren verfügen stets über die höchsten Berechtigungen, während Bediener nur über eingeschränkte Berechtigungen verfügen.
7. Ruida Packing Machinery: Bereitstellung von FDA- und GMP-konformen Maschinen
Manchmal sind Hersteller zwar bereit, ihre Maschinen mit einem Audit-Trail-System auszustatten, doch nicht alle Maschinen unterstützen dies. Ruida Packing arbeitet mit namhaften Pharmaunternehmen wie UCB und US Pharma zusammen und hat sich zweifellos der Entwicklung von Teil-11-konformen Anlagen verschrieben. Unsere Maschinen verfügen über CE-, cGMP-, ISO- und weitere relevante Zertifikate. Je nach Kundenbedarf kann Ruida auch Überwachungslösungen gemäß 21 CFR Part 11 für Produktions- und Verpackungsanlagen bereitstellen.
Ruida unterstützt die pharmazeutische Fertigung zudem mit praktischen Serviceleistungen:
Zertifizierte Materialien: Edelstahl 304 für berührungsfreie Teile und Edelstahl 316L für kontaktbehaftete Teile.
Inbetriebnahme-Services per Fernzugriff und vor Ort, mit Kundendienstzentren in Nordamerika und Hongkong.
Umfassende Maschinenanleitung: Handbücher, Bedienungsanleitungen, Wartungsunterlagen und Kundendienstvideos.
IQ-, OQ- und PQ-Dokumentation: Optionale Unterstützung bei Validierungsunterlagen, IQ, OQ und PQ sowie bei der Erstellung von Konformitätsunterlagen.
Für Hersteller, die die Einhaltung von 21 CFR Part 11 planen, kann die Wahl von Anlagen mit konformitätsgerechten Optionen zukünftige Umrüstungskosten senken und die Vorbereitung auf Audits erleichtern.
8. Fazit
Wenn wir zurückblicken, wann 21 CFR Part 11 in Kraft trat, können wir erkennen, wie tiefgreifend diese Vorschrift die Branche seit 1997 geprägt hat. Seit dem 20. August dieses Jahres ist diese Vorschrift der Maßstab dafür, wie von der FDA regulierte Unternehmen elektronische Daten verwalten sollten. Heutzutage ist die Einhaltung von 21 CFR Part 11 ein Schwerpunkt und ein Anliegen für viele Pharma- und Nutra-Hersteller, und Sie sollten sich besser darauf vorbereiten. Nicht nur die Validierung, sondern auch die Maschinen.
9. Häufig gestellte Fragen
Frage 1: Ist 21 CFR Part 11 verbindlich?
Ja, natürlich. Sie gilt jedoch nicht für alle elektronischen Aufzeichnungssysteme. Sie ist verbindlich, wenn elektronische Aufzeichnungen oder elektronische Signaturen verwendet werden, um die Anforderungen der FDA zu erfüllen.
F2: Worin besteht der Unterschied zwischen GMP und 21 CFR Part 11?
Nun, sie haben unterschiedliche Schwerpunkte. GMP konzentriert sich auf die Produktqualität, während 21 CFR Part 11 den Fokus auf elektronische Aufzeichnungen und elektronische Signaturen legt.
F3: Muss jede pharmazeutische Maschine Part-11-konform sein?
Nein. Eine Maschine muss im Hinblick auf Part 11 berücksichtigt werden, wenn sie regulierte elektronische Aufzeichnungen erstellt, speichert, ändert oder überträgt. Rein mechanische Anlagen erfordern möglicherweise nicht denselben Kontrollgrad.
F4: Was ist ein Prüfpfad?
Ein Prüfpfad dient dazu, alle im System durchgeführten Aktionen aufzuzeichnen und dabei die genaue Aktion, den Zeitpunkt sowie etwaige Änderungen anzuzeigen.
F5: Woran erkenne ich, ob eine Anlage die Anforderungen von Part 11 erfüllt?
Sie können prüfen, ob sie Audit-Trails, Benutzerzugriffskontrolle, elektronische Signaturen, sichere Datenspeicherung, Datenexport und Validierungsdokumente unterstützt. Aber um es einfach zu sagen: Fragen Sie vor dem Kauf einfach den Maschinenlieferanten.
[1] US-amerikanische Food and Drug Administration: Part 11, Elektronische Aufzeichnungen; Elektronische Signaturen – Geltungsbereich und Anwendung
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/part-11-electronic-records-electronic-signatures-scope-and-application
[2] Electronic Code of Federal Regulations: 21 CFR Teil 11 – Elektronische Aufzeichnungen; Elektronische Signaturen
[3] Federal Register: Elektronische Aufzeichnungen; Elektronische Signaturen – Endgültige Regelung, veröffentlicht am 20. März 1997